
Почему нам важна пространственная структура белка, какие существуют повторяющиеся паттерны и как нейронные сети могут помочь решить вопросы биоинформатики — эти темы рассмотрим в нашей статье.

На картинке мы видим белок, который тащит за собой что-то, похожее на коронавирус, но это, конечно, не он. Белок шагает по другой структуре при помощи «ног». Нужно понимать, что это не фотография и не реальное видео, а просто визуализация – сфотографировать белок мы пока не можем. Белки – самые маленькие структуры, которые нам известны, однако при этом они занимаются очень многим: они умеют ходить, цепляться за другие структуры, находить какие-то специальные объекты, например, вирусы в нашем организме.
Вообще белки выполняют самые разные функции – они являются очень важной частью иммунной системы, и, по сути, представляют собой антитела. Большое количество белков содержатся в слюне, они помогают нам с пищеварением и делят глюкозу и другие длинные органические молекулы на кусочки, чтобы их было проще переваривать. Белки составляют молекулы гемоглобина – у нас есть гемоглобиновые клетки, которые переносят кислород в крови, и внутри этих клеток как раз содержится очень много белка гемоглобина. Таким образом, гемоглобин устроен специально так, чтобы он мог переносить на себе большое количество кислорода, поэтому обеспечение клеток кислородом осуществляется именно при помощи белков.
Нам известно около 200 миллионов белков, на нашей планете они составляют основу жизни. Вся жизнь на Земле имеет белковую структуру, даже вирусы, и пусть вирусы не называют жизнью, они все равно в некотором смысле живые.
Пространственная структура белка
То, как этот белок шагает, вызвано исключительно его химической и физической структурой. Его вращения вызываются химической реакцией, но также на его передвижение воздействуют силы на уровне атомов.
Почему же нам так важна пространственная структура? Потому что именно она в большой степени определяет функциональность белка. Если мы хотим понять, как именно гемоглобин переносит на себе кислород, то, по сути, нам нужно узнать, каким образом он сворачивается в трехмерном пространстве так, что в него хорошо упаковывается кислород. Здесь вы видите, что у белка есть специальные клешни, которые взаимодействуют с круглой структурой сверху, а у нее, в свою очередь, есть белки на поверхности, которые «цепляются» за держатель.
О пространственной структуре белков узнать что-либо очень сложно, так как мы еще не можем смотреть на такие маленькие размерности. Например, чтобы придумать лекарство, мы должны рассматривать химические структуры на размерности 0,3 ангстрема, ангстрем – 10 -10 метра.
Casp и нейронные сети
Сегодня будем фокусироваться на последних достижениях компании Alphabet от Google и её лондонского подразделения DeepMind. В 2018 и 2020 году на соревнованиях Casp они побили рекорды других алгоритмов, основанных на нейронных сетях.

На рисунке вы видите результаты этого соревнования – вертикаль показывает качество алгоритма. Видно, что Alphafold 2020 обладает качеством выше 80, хотя все предыдущие алгоритмы не получали значения выше 40. То есть, достижение 2020 года для науки просто огромно. Об этом говорилось во множестве научных журналов, таких как Nature, Science, об этом писали и в популярных журналах, типа Forbes или Fortune – настолько все были потрясены новостью.
Структура белков

На рисунке вы видите два белка, их названия и функциональность нам не так важны, как пространственная структура. Есть несколько способов ее отображения. Здесь представлен «ленточный» способ – так мы лучше видим, какие поверхности образуются в белках во время сворачивания. На рисунках использованы два цвета, синим обозначено то, что предсказывает алгоритм. Стоит отметить, что это не абсолютная биологическая истина, а всего лишь попытка к ней приблизиться.
Синие поверхности очень хорошо отражают зеленые – при этом, между ними все-таки есть расстояние, на рисунке это заметно. Также можно заметить, что у структур есть некие повторяющиеся паттерны:
- Спиралька, или alpha helix.
- Параллельные друг другу ленты, или beta sheet.
Как образуются белки
В нашем организме протеины образуются из аминокислот – прикрепление аминокислот к друг другу происходит путем отбрасывания наконечника аминокислоты. Протеины же выглядят как цепочка – они не обладают разветвлениями.
Как только цепочки образуются, то сразу начинают сворачиваться, так как в них действует большое количество химических сил. Они сворачиваются в alpha helix или beta sheet, так как не могут противостоять внутриатомным и внутримолекулярным воздействиям. После этого они начинают сворачиваться в более сложные структуры, которые называются структурами третьего порядка. Не все белки третьего порядка имеют какую-то одинаковую структуру: они могут быть плотными и растянутыми, но от плотности зависит их функциональность. Плотные структуры могут что-то держать или проникать в другие виды структур.
ДНК и РНК
Все начинается с ДНК, которая хранится в ядре клетки. Люди – эукариотические существа, это значит, что наша ДНК хранится внутри плотной оболочки, то есть ядра. Это важно, так как ДНК выступает в роли банка, где хранится то, что должно оставаться защищенным от любых воздействий, в особенности, от вирусов. Все, что присутствует в нашем организме образуется путем чтения ДНК и генерации белков по тому, что было прочитано.
Но так было не всегда: считается, что жизнь началась не с ДНК, а с РНК, и первый миллиард лет существования жизни на земле был этап применения РНК, то есть все одноклеточные существа строились на рибонуклеиновой кислоте.

РНК отличается от ДНК тем, что в ней вместо двух последовательностей записана только одна. РНК строится почти на тех же компонентах, что и ДНК, за одним исключением – ДНК состоит из четырех букв или четырех базовых элементов: тимин, цитозин, аденин и гуанин. Они связаны друг с другом и кроме того, связаны попарно – тимин, например, может соединяться только с аденином, а цитозин с гуанином. В РНК они не могут соединяться попарно, да и вообще прикрепляться друг к другу, кроме того, вместо тимина в РНК присутствует урацил.
В мире до ДНК существовала одна проблема – как только внутри РНК происходили какие-то изменения, например, в цепочку включался вирус и заменял последовательность или в результате мутации в структуре что-то ломалось, то первоначальный вариант становился уже недоступен, так как существовала только одна копия. Абсолютное большинство мутаций – вредные, так что защищать информационную безопасность организма - это важная задача, именно поэтому самые прогрессивные организмы после первого миллиарда существования перешли на использование ДНК.
ДНК и РНК очень тесно связаны.

На картинке вы видите транскрипцию ДНК в РНК. ДНК хранится только в ядре – это самое защищенное место в клетке, так как в ядро может попасть только определенное количество допущенных туда элементов типа РНК-полимеразы. РНК-полимеразы разделяют две части ДНК, берут одну часть и транслируют ее в РНК, пользуясь тем фактом, что цепочки ДНК попарны. Тимин объединяется с аденином, а аденин с урацилом, цитозин с гуанином, а гуанин с цитозином, то есть ДНК напрямую транслируется в РНК. Трансляций в день происходит большое количество, и они могут происходить неправильно. Но каждая «неправильность» будет непохожа на другую – таким образом, получится случайная мутация, что в целом, безопасно для нашего организма, так как наша иммунная система отлично борется с такими проблемами.

РНК делится на комеры или тримеры. Каждые три последовательные буквы в РНК соответствуют определенной аминокислоте.
Как же из РНК получаются белки?
Есть другой тип полимеразы, который крепится снаружи ядра к РНК и по три буквы транслирует один алфавит, состоящий из четырех букв в другой алфавит, состоящий из двадцати букв. На рисунке представлены аминокислоты, на которых построена вся жизнь, которую мы знаем, и их химические формулы. Это достаточно простые конструкции, и кроме того, очень похожие – у всех присутствует ОН окончание, а в середине есть перемычка, которую мы потом будем использовать как точку отсчета для того, чтобы высчитывать трехмерную структуру белков. Все аминокислоты могут объединяться друг с другом.

Q: Что означает звездочка внизу?
А: Это стоп-кодон, который означает начало или конец последовательности. Например, у бактерии кишечной палочки, как и у всех бактерий, геномная последовательность записана кольцевым способом, поэтому важно понимать, где начало, где конец.
Итого: у нас есть два четырехбуквенных алфавита – ДНКовый и РНКовый – и один двадцатибуквенный, из которого состоят белки. Белки бывают разной длины. На рисунке ниже вы видите гистограммы распределения белков в нашем организме:

Как видите, большая часть белков содержит в себе около 200-300 аминокислот. Каждая аминокислота – это химическая молекула, сложная химическая конструкция и наших вычислительных способностей, чтобы её рассчитать, пока не хватает. Эта трехмерная структура обладает несколькими свойствами – она начинает появляться тогда, когда мы начинаем транслировать РНК в белковую последовательность.
Есть два основных момента, о которых нужно помнить – это притяжение и отталкивание, которые объясняют то, какая в итоге получается структура. Процесс сворачивания белка обычно занимает миллисекунды, но бывают и исключения – микросекунды или даже дни.
Структура на картинке ниже обладает минимумом с точки зрения энтропии. Молекула сворачивается из-за того, что на неё действуют силы притяжения и отталкивания, и потому что ей нужно минимизировать ту энергию, которую она в себе несёт. Пространство, которое вы видите на рисунке справа, описывает энтропию этой конкретной молекулы белка. Казалось бы, можно просто посчитать минимальную энтропию и дело с концом. Но есть так называемый парадокс Левенталя, который посчитал возможное количество возможных сворачиваний каждой белковой молекулы. У него получилось цифра 10143 , что очень сильно превосходит количество атомов во вселенной, и, соответственно, у нас просто не хватит времени жизни, чтобы произвести такие расчеты.
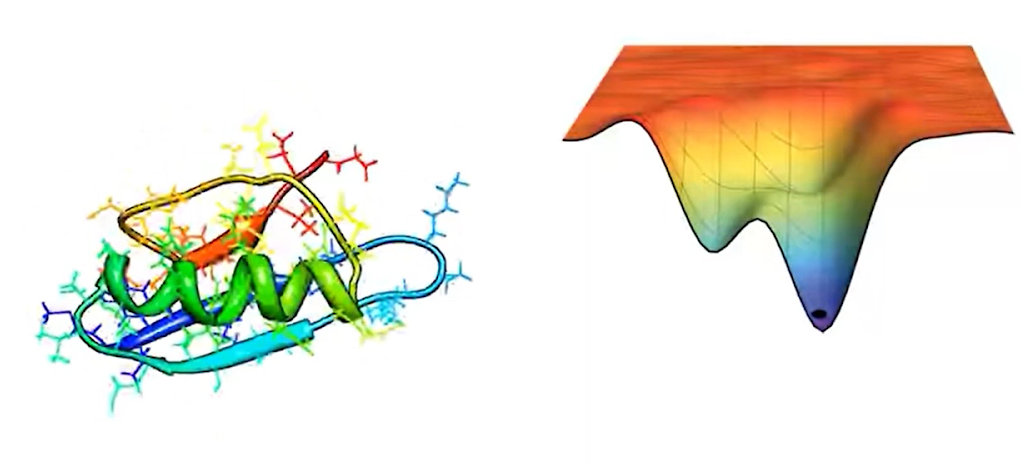

Здесь вы видите практически то же самое: не свернутая молекула обладает огромной энтропией и не может долго оставаться в таком виде, поэтому внутри себя она переходит в нативный state, где остается очень долгое время.
Существует один или пять процентов белков, которые не могут так просто переходить в нативный state. Известно, что на нативный state влияет внешняя среда, а именно температура, раствор, соседи и другие факторы.
Рентгеновская кристаллография
Задачу по структуре белка решают уже 60 лет с помощью самых разных способов, один из которых называется рентгеновская кристаллография.

Метод заключается в том, что вокруг какой-либо наживки наращивается кристаллическая структура того белка, про который мы хотим что-то узнать. После того, как кристалл вырастает до определенных размеров, мы светим на него в разных проекциях, рассматриваем получившийся паттерн и пытаемся подобрать такую пространственную структуру, которая объяснила бы паттерны в тех нескольких проекциях, которые мы видим. У нас получается некая модель, которую мы снова накладываем на паттерн – таким образом мы выводим какие-то данные. Точность метода относят к 95 процентам.
За 60 лет рентгеновской кристаллографией и другими методами экспериментального характера была посчитана пространственная структура 170.000 белков из 200.000.000 известных на земле. Эксперименты долгие, сложные и дорогие, поэтому в качестве объектов выбирались самые важные в биологическом смысле белки – те, которые находятся в теле человека, и белки из модельных организмов типа кишечной палочки.
Вычислительные методы
Как только появилась биоинформатика, люди стали пытаться решить эти задачи вычислительным путем, применяя различные алгоритмы.

Есть организация под названием Folding Net Home, которая представляет собой распределенные вычисления. Можно зайти к ним на сайт, скачать определенное программное обеспечение, которое будет рассчитывать на вашем компьютере трехмерную структуру белков. Сеть действует вот уже 20 лет, с начала нашего века. В апреле 2020 года к сети было подключено огромное число новых машин, чтобы понять принцип работы белка нового коронавируса, а именно того спайка, который протыкает мембрану наших клеток.

Именно тогда распределенная сеть совершила большой скачок мощности и обошла все имевшиеся на тот момент суперкомпьютеры.

Алгоритмы, которыми Folding Net Home считает структуру белков, построен на марковских сетях, и его основная задача – предсказать трехмерную структуру молекул. Точность этого алгоритма по сравнению с современными методами не очень высока, поэтому скорее всего, сеть поменяет зону своей применимости на другие вычислительные задачи.
Q: Получается, Folding Net Home – это опен-сорсное решение, и к ней может получить доступ каждый?
А: Я не смотрела точно специфику, но знаю, что это точно не коммерческое решение. И Alphafold тоже сейчас открытый.
Q: Зачем нам вообще знать трехмерную структуру белка?
А: Она дает нам понимание того, как, собственно, работает белок. То есть, к чему он умеет прикрепляться, что он умеет на себе носить или что расщепляет. Это необходимо для изобретения новых лекарств. То есть вместо того, чтобы случайным образом найти то, что сработает в качестве лекарства, мы можем все рассчитать заранее. Берем коронавирус, он состоит из нескольких белков на его поверхности, находим что-то, что хорошо умеет к нему прикрепляться и легко выводится из организма, и таким образом получаем идеальное лекарство. Сейчас, кроме лекарств, с помощью Alphafold находят белки, которые хорошо умеют расщеплять пластик. И можно, зная пластик, сконструировать несуществующий пока белок, который обладал бы такой пространственной структурой, чтобы его можно было бы разложить. Этими исследованиями занимаются уже давно, но активное развитие они получили в 2021 году.
Q: Вы говорили, что сворачивание зависит от окружающей среды. Сейчас человечество активно осваивает космос, где нас ждут совершенно другие условия. Как это будет влиять на изучение белков в дальнейшем?
А: Скорее всего, повлияет очень сильно, но пока трудно проводить такие масштабные исследования. Мы даже не знаем, какой процент молекул может менять свой нативный state, исходя из окружающей среды. Кроме того, все alpha helix, которые есть на нашей планете закручены в правую сторону, и совершенно нет тех, что были бы закручены влево. Возможно, что на какой-то планете будет наоборот.
Alphafold и Casp
Casp поделен на простые, средние и сложные кейсы, большая часть из которых получена с помощью рентгеновской кристаллографии, небольшая часть – поляризованным светом или раствором. Соревнование Casp проводится каждые два года.
Вспомним, что у каждой аминокислоты есть условный центр, который называется бета-углерод или альфа-углерод.
Также есть несколько вариантов кодирования – первый, самый очевидный, это координация в трехмерном пространстве. У него есть минусы, так как мы можем переворачивать свернутый белок бесконечное число раз и при этом получать разные результаты. Можно пользоваться парными комбинациями между буквами и последовательностью углов между ними. Здесь тоже есть проблема, так как последовательность углов сама по себе неустойчива – небольшое изменение угла влечет за собой и изменение трехмерного белка.

Q: Мы предсказываем расстояние для каждой точки?
А: Для каждого центрального атома углерода в аминокислоте.
Данные на вход
Очевидно, что на вход нужно положить буквенную последовательность аминокислот. Если длина последовательности аминокислот – 200, то размер таблички будет 200 на 20, и в каждой из 200 колонок будут нули кроме 1. Последовательность передается сразу целиком.
Несмотря на то, что в нашем мире существует огромное количество живых организмов, все они устроены очень похоже, и их белки практически одинаковы. Поэтому если мы посчитали структуру для какого-то млекопитающего, то эти данные тоже можно положить на вход. Также на вход цепляют так называемые шаблоны – белки, похожие на входной, с уже известной структурой.
Белки постоянно мутируют. Чаще всего выживают организмы, где мутации не слишком повлияли на жизнеспособность. Есть кусочки белка, которые всегда остаются неизменными, поэтому эти эволюционные моменты также могут подсказать нам структуру белка.
Также стоит задуматься о коэволюции: если в важных местах что-то все-таки мутирует, то обычно оно мутирует попарно.

Вся таблица, представленная выше, называется MSA и тоже подается на вход.
У нас есть входной протеин, в нем 21 буква и наша таблица, а также шаблоны похожих белков. Каждая штука специально клипается, в этом заключается её обработка. Здесь получаем квадрат 256 на 256.

Итого у нас получилось 6 массивов. Мы подаем их на вход в AlphaFold.

Первое, что пытаемся сделать – подсчитать имбединги для MSA и записать попарные взаимодействия внутри белка.

Следующее подается на вход в Eva Former, это трансформер.

Получившееся мы подаем в структурный модуль: еще один модуль нейронной сети, который позволяет ему на нескольких стадиях читать лоссы. Сперва мы получаем какое-то грубое представление про гистограмму и про углы, затем мы рефайним их и считаем лоссы, в итоге получаем трехмерные координаты всех атомов.

Q&A
Q: Какая метрика у самих лоссов?
А: Лосс регрессионный, но построено все так, чтобы он был инвариантен по отношению к системе координат.
Стоит сказать, что AlphaFold применяется итеративно.
Для всех протеинов длиной 1400 применяется инференс на одной GPU.

Команда AlphaFold уделила большое внимание спайку коронавируса, что помогло появлению новых лекарств.
Q: Какого порядка должны быть условия изучения, чтобы исследование лекарств считалось безопасным?
А: Для того, чтобы делать лекарства нужно знать структуру белка с точностью до 0.3 ангстрема. Alphafold предсказывает больше половины предложенных белков.
Q: То есть мы просто должны выучить еще большую модель, которая будет предсказывать данные независимо от домена?
А: Скорее всего, зависимость от домена все же будет, но цель именно в том, чтобы можно было заинферить какой-нибудь белок, даже придуманный тобой, и посмотреть, как он будет выглядеть в пространстве.
Q: Не получается ли, что вопрос качества еще открыт, так как методы все же не точны?
А: Я так понимаю, что самый классный способ избавиться от bias – это прогнать один и тот же белок через несколько разных экспериментальных способов получения пространственной структуры и сравнить с тем, что делает Alpha Fold. Это как раз и делали для белка ковида и получилось весьма неплохо, однако есть куда расти.
Q: А имбединг как-то пытались проанализировать? Обычно же сравнивают со словами-синонимами и по семантике?
А: Думаю, что если кто-то это и делал, то сейчас к полученным результатам у нас нет доступа, но мне кажется, что статьи появятся достаточно скоро.
По материалам открытого семинара Анны Петровичевой «Нейронные сети для распознавания пространственной структуры белков», который прошел в Xperience.AI.


